Pooled CRISPR functional genomics screens are now among the most powerful methods available for genome-scale gene function mapping, genetic interaction discovery, and therapeutic target identification. Every such screen depends on a single upstream deliverable: a sgRNA library that is sequence-accurate, uniformly represented, and correctly constructed. When that library is compromised — by synthesis bias, PCR-induced skew, insufficient cloning coverage, or imprecise titer estimation — the compromise propagates invisibly through every subsequent experimental step and surfaces as uninterpretable screen noise only after weeks of work.
This guide covers the complete workflow from computational guide design through oligo pool synthesis, PCR amplification, cloning, quality control, lentiviral production, and screen execution. The emphasis throughout is on the specific decisions — not generic principles — that determine whether a library will support publication-quality screen data or require troubleshooting that consumes the timeline of the project.
Why Oligo Pools Are the Standard Format for sgRNA Library Construction
A genome-wide CRISPR library targeting all human protein-coding genes requires between 60,000 and 120,000 unique guide sequences, depending on the number of guides per gene. A focused pathway or disease-relevant gene set library may require 2,000 to 20,000. Neither range is feasible through one-by-one synthesis: cost, logistics, and turnaround time all make individual column-based synthesis impractical at these scales.
Array-based oligo pool synthesis solves this by producing all designed sequences in parallel on a single chip in one production run. Dynegene's Oligo Pools synthesis platform supports up to 4.35 million unique sequences per chip, at oligo lengths of 2 to 350 nt, generating up to 1 Gb of synthesized DNA per run. For a standard genome-wide sgRNA library requiring 80,000 unique 80 nt oligos, this represents a single chip occupying less than 2% of the available synthesis capacity — meaning the same production run can accommodate a complete library with room for internal controls, spike-in sequences, and validation guides at no additional synthesis step.
Beyond capacity, oligo pool synthesis provides complete design flexibility. The guide sequences, flanking architecture, control composition, and library complexity are entirely user-specified. Unlike pre-built commercial libraries where content is fixed, a custom oligo pool-based library can be designed to target specific isoforms, non-coding regulatory elements, therapeutic targets not yet represented in commercial offerings, or any other genomic features relevant to the scientific question.
Step 1: Computational sgRNA Design
The quality ceiling of a CRISPR screen is set at the guide design stage. Poor design — low on-target activity, high off-target potential, inadequate guide coverage per gene — cannot be compensated for by any optimization downstream. This step deserves more analytical investment than is typically allocated.
PAM Site Identification and Nuclease Compatibility
Guide sequences must be adjacent to a protospacer adjacent motif (PAM) recognized by the Cas nuclease being used. For Streptococcus pyogenes Cas9 (SpCas9), the canonical PAM is NGG on the non-template strand — the 20 nt immediately 5′ of this NGG constitutes the targeting sequence. For SaCas9, the PAM is NNGRRT. For AsCas12a (Cpf1), the PAM is TTTN located 5′ of the protospacer rather than 3′. For engineered SpCas9 variants such as SpRY, near-PAMless activity enables targeting positions unavailable to wild-type SpCas9, but activity remains position-dependent.
Guide design must begin with accurate PAM site identification across the full target region set. PAM sites missed during this step represent guides that will not direct productive Cas9 binding regardless of sequence quality.
On-Target Activity Scoring: Specific Sequence Determinants
Position-specific sequence preferences significantly influence editing outcomes, and these preferences are not captured by overall GC content alone. According to published mechanistic studies and validated scoring algorithms:
- GC content in the PAM-proximal seed region (positions 1–12 from the PAM) should be maintained at 45–55%. GC content exceeding 56% in this region impairs Cas9 conformational dynamics, reducing cleavage efficiency by up to 40%.
- GC content in the PAM-distal region (positions 13–20) of 55–65% enhances RNA-DNA hybridization stability and improves functional outcomes by approximately 1.8-fold in genome-wide applications.
- Guanine at position 20 (adjacent to the PAM) demonstrates superior performance over cytosine at the same position.
- Homopolymer runs of 4 or more consecutive identical nucleotides should be avoided — these impair both synthesis fidelity and Cas9 recognition.
- Overall guide GC content should be in the 40–60% range.
The following specifications serve as a design quality threshold table:
| Design Parameter |
Optimal Specification |
Acceptable Range |
Rationale |
| Guide sequence length |
20 nt |
19–21 nt |
Balances specificity and efficiency |
| Overall GC content |
50% |
40–60% |
Maintains thermal stability |
| PAM-proximal GC (nt 1–12) |
45–55% |
40–56% |
Prevents Cas9 conformational impairment |
| PAM-distal GC (nt 13–20) |
55–65% |
50–70% |
Enhances RNA-DNA hybridization |
| Maximum homopolymer run |
0 preferred |
≤4 consecutive bases |
Ensures synthesis fidelity |
| Off-target sites (genome-wide) |
0 perfect matches |
≤2 sites with ≥3 mismatches |
Maintains screen specificity |
| Guides per gene (pooled screen) |
6 independent sgRNAs |
4–8 per target |
Reduces false-positive identification rate |
| Non-targeting controls |
100–500 guides |
≥1% of library size |
Establishes baseline variation |
Several well-validated scoring algorithms are available: Rule Set 2 (Doench et al., 2016), DeepCRISPR, and CRISPRscan each provide complementary predictions based on different training datasets and model architectures. Applying two algorithms and prioritizing guides that rank consistently in the top quartile of both is more robust than relying on a single tool.
Off-Target Prediction and Filtering
Off-target cutting confounds phenotypic screens because any phenotype associated with a guide that cuts outside its intended target cannot be unambiguously attributed to the on-target gene. Filtering off-target-prone guides before synthesis is the correct intervention; attempting to attribute off-target effects in screen data analysis is not reliable.
Tools including Cas-OFFinder, CRISPOR, and Bowtie-based alignment pipelines provide genome-wide specificity assessment. The standard filtering criterion is exclusion of guides with any predicted off-target site with fewer than 3 mismatches. Including 4 to 8 independent guides per gene provides the statistical redundancy needed to distinguish genuine on-target hits from off-target artifacts.
Guide Coverage per Gene and Control Composition
Most published genome-wide libraries include 4 to 6 guides per gene. For focused libraries where statistical power per target matters more than breadth, 6 to 8 guides per gene is appropriate. Positive controls should target genes with well-characterized, reproducible phenotypes in the assay model. Negative controls should comprise 1% or more of the total library, composed of non-targeting sequences or guides against genes confirmed to have no effect on the assay readout. Control representation must be specified in the design file and maintained at that level through synthesis to enable accurate normalization during data analysis.
Step 2: Oligo Architecture Design for Synthesis
The guide sequences selected computationally are not submitted directly to synthesis. Each oligo in the pool must carry all functional elements required for downstream cloning, in the correct order and with the correct lengths.
Standard Oligo Architecture
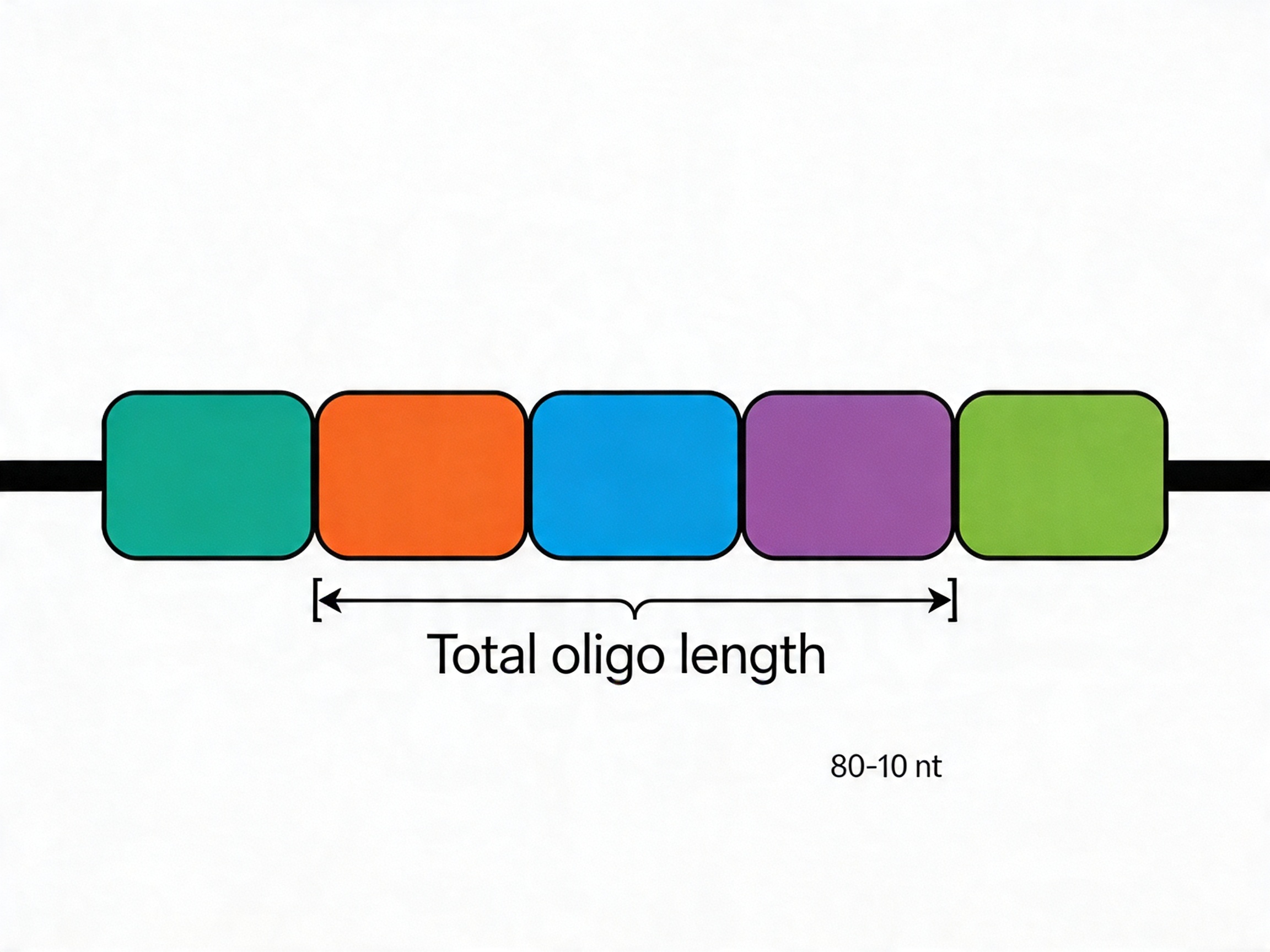
A standard CRISPR library oligo for a BbsI or BsmBI Golden Gate cloning strategy includes:
- 5′ universal primer binding site (18–22 nt): Identical across all oligos; used for PCR amplification of the pool
- Optional unique barcode (10–20 nt): Required for multiplexed screening applications; designed with minimum Hamming distance ≥ 2–3 nt between any pair
- 20 nt protospacer sequence: The variable targeting element; unique per oligo
- Partial sgRNA scaffold or direct cloning overhang: Compatible with the specific vector's cloning site
- 3′ universal primer binding site (18–22 nt): Reverse complement of the reverse PCR primer; identical across all oligos
Internal Restriction Site Removal
Oligos must be screened computationally for internal occurrences of the restriction enzyme recognition sites used in cloning. For Golden Gate strategies using BbsI (recognition: GAAGAC) or BsmBI/Esp3I (recognition: CGTCTC), any internal occurrence of these sequences within the protospacer or flanking elements will cause incomplete or mis-directed digestion of the amplified library. For ligase-based strategies, EcoRI, BamHI, NotI, BglII, and other common cloning enzymes should also be screened. When a collision is found, silent substitutions at wobble positions (third codon position) can resolve the conflict without changing the targeting sequence's functional identity.
Barcode Design for Multiplexed Applications
For screens requiring unique construct identification — dual-guide combinatorial libraries, multiplexed barcoded screens, or molecular recording applications — barcode sequences require design with minimum Hamming distance specifications ensuring unambiguous identification even after sequencing errors. Standard 10 nt barcodes with minimum 2–3 nt Hamming distance between any pair tolerate synthesis and sequencing error rates while maintaining correct demultiplexing. For applications tolerating up to 3 base mismatches or indels, sequences differing by 4 or more nucleotides provide robust discrimination.
Step 3: Pool Synthesis and Initial Quality Assessment
Dynegene's CRISPR sgRNA Library service provides synthesis of sgRNA libraries using the same microarray-based synthesis platform as the oligo pools service. Upon receipt of the synthesized pool, confirm the following before proceeding to amplification:
- Yield: Verify that mass is sufficient for planned amplification reactions. Most library PCR workflows require 1–10 ng input per 50 µL reaction.
- Size distribution: Capillary electrophoresis or Bioanalyzer confirmation of the expected size peak and absence of major truncation products.
- Representation check (recommended for large libraries): A shallow sequencing run (1–5 million reads) of the raw pool, mapped to the designed guide list, confirms pre-amplification representation uniformity. Any systematic skew identified at this stage reflects synthesis rather than downstream processing and can inform amplification strategy.
Dynegene's platform specifications document accuracy and uniformity as core performance requirements — not secondary metrics. Representation skew introduced at the synthesis stage will be amplified, not corrected, by every subsequent processing step.
Step 4: PCR Amplification
PCR amplification converts the single-stranded pool into double-stranded amplicons with defined termini ready for restriction digestion and cloning. This step is the primary source of representation skew in library construction beyond the synthesis pool itself.
Cycle Number and Polymerase
Restrict amplification to 15–20 total cycles. Beyond this range, sequences with favorable primer binding kinetics or reduced secondary structure are enriched at the expense of others, producing skew that cannot be corrected later. Skew ratio (90th percentile / 10th percentile of guide abundance) should remain below 2.0 in well-optimized amplification; poorly controlled amplification protocols exhibit skew ratios above 5.0, which require 10-fold higher cellular coverage in the screen to compensate.
Use a high-fidelity proofreading polymerase with error rate below 1×10⁶ per base per duplication — Q5, Phusion, or equivalent enzymes are appropriate. KAPA HiFi or Phusion GC Buffer formulations improve uniformity for pools with GC-extreme guide sequences.
For sequences with GC content above 70% or below 30%, additives that address differential amplification efficiency may be required: betaine at 1–2 M final concentration and DMSO at 2–5% v/v are the most commonly used.
Reaction Setup for Representation Preservation
Run 4 to 8 parallel reactions from the same pool aliquot and pool the products before cleanup. This reduces the risk that stochastic early-cycle differences in any single reaction dominate the final product. Primer concentration should be 0.2–0.5 µM and annealing temperature should be set 2–3°C below the calculated Tm of the universal primers.
Step 5: Cloning into the sgRNA Expression Vector
The amplified library insert is digested and cloned into the sgRNA expression vector. Most lentiviral CRISPR vectors use a U6 promoter for guide expression and include a selection marker and fluorescent reporter for monitoring transduction.
Golden Gate Assembly for Large Libraries
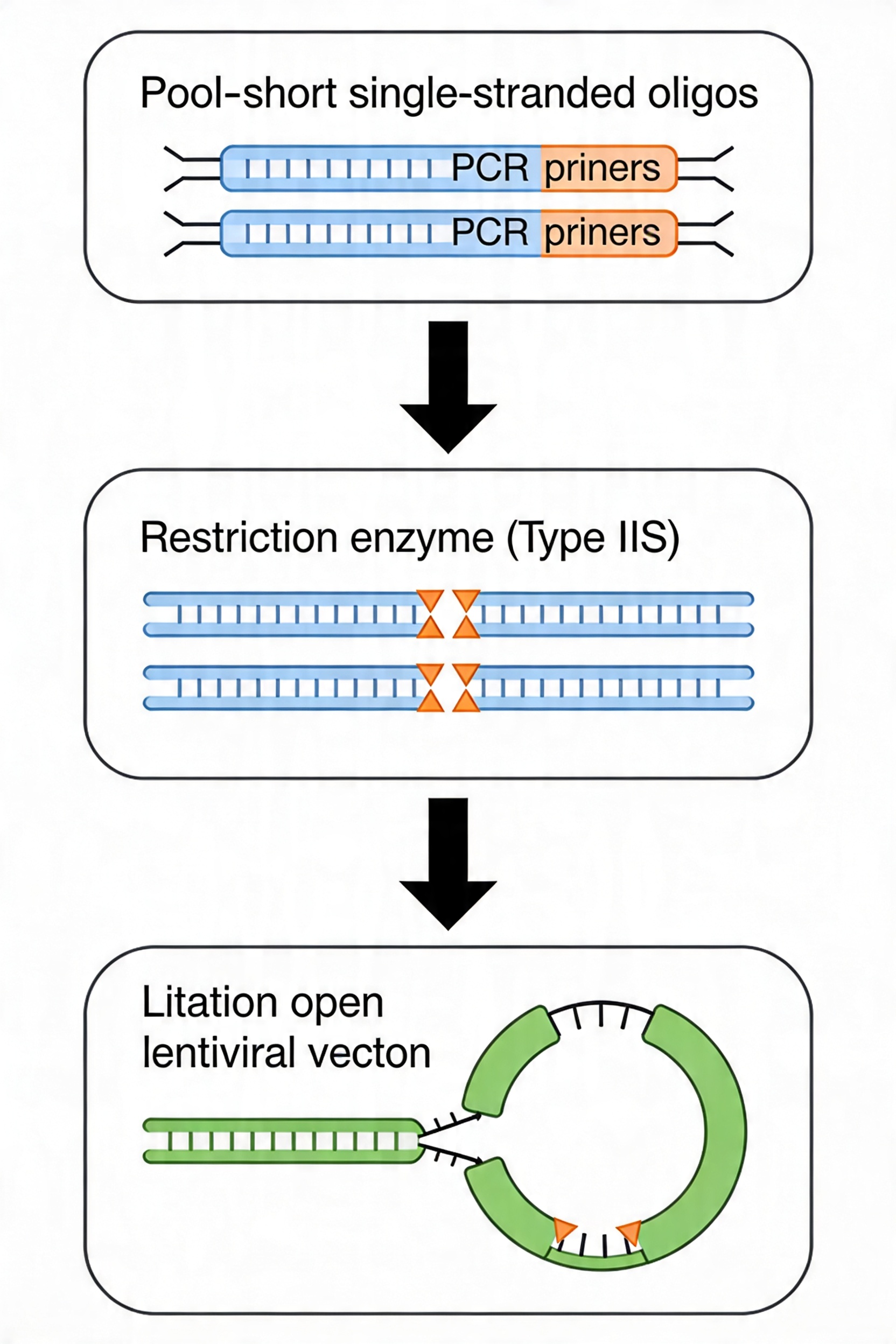
Golden Gate Assembly using Type IIS restriction enzymes (BsmBI/Esp3I or BbsI) is the preferred cloning strategy for libraries above approximately 10,000 guides. BsmBI generates 4-nt overhangs by cutting 1 nt 3′ of its recognition sequence on one strand and 5 nt 3′ on the other; these 4-nt overhangs are sequence-defined by the guide sequence termini and the vector backbone, enabling directional ligation without random ligation between non-adjacent fragments. Combining restriction and ligation into a single-tube cycling reaction (typically 25–30 alternating cycles of digestion at 37°C and ligation at 16°C) improves efficiency over sequential digest-and-ligate protocols.
For overhang design in combinatorial multi-guide libraries: GC content of 45–60% with Tm of 60–65°C, secondary structure free energy less than –3 kcal/mol, absence of restriction recognition sequences, and minimum 5 mismatches compared to all other overhangs in the assembly are the validated design criteria.
Transformation Coverage Requirements
Colony count is the most commonly underestimated parameter in library cloning. The minimum acceptable coverage is 100-fold the library size — for an 80,000-guide library, this means 8 million independent colonies. The recommended target for optimal representation uniformity is 500 to 1,000-fold coverage, or 40–80 million colonies for the same library size.
Achieving this requires electrocompetent cells prepared to ≥10¹⁰ CFU/µg, multiple parallel electroporations (4 to 8 cuvettes) to reduce arcing risk while increasing aggregate efficiency, and plating on large-format agar (25 cm × 25 cm) plates. Recovery in pre-warmed SOC medium for 60–90 minutes before plating, and incubation at 30°C rather than 37°C for 16–20 hours, reduce metabolic selection bias and improve colony uniformity.
Colony harvesting must be performed by scraping the entire plate surface — not by picking individual colonies — to capture all library members without selective sampling.
Step 6: Plasmid Library Quality Control by NGS
Before lentiviral production, the plasmid library must be quantitatively validated by deep sequencing. Amplify the guide insert region from the plasmid pool using primers flanking the cloning site. Prepare a sequencing library with Illumina adapters and sequence to a depth providing a minimum of 200 to 500 reads per library member.
Key QC Metrics
From the sequencing data, calculate:
- Guide detection rate: The percentage of designed guides present at ≥1 read. A high-quality library should detect ≥95% of designed guides.
- Skew ratio (90/10): The ratio of 90th percentile guide count to 10th percentile guide count. Target below 2.0; values above 5.0 indicate problematic representation and require protocol review before proceeding.
- Percent guides within 10-fold of median: The fraction of guides with abundance between 0.1× and 10× the median.
- Control guide representation: Verify that positive and negative controls are present at expected relative abundance. Systematic dropout of controls at this stage indicates a cloning or propagation problem.
Sequencing Platform Considerations
Illumina two-color chemistry platforms (NextSeq, NovaSeq) exhibit signal artifacts with polyguanine sequences due to the absence of a G-channel signal. Four-color chemistry platforms (MiSeq, HiSeq 2500) perform better for homopolymer-containing sequences common in some sgRNA designs.
Paired-end sequencing (2×150 bp or 2×250 bp) enables detection of synthesis errors and PCR artifacts in the read overlap region. Quality score filtering at Phred Q30 minimum (99.9% base call accuracy) is appropriate for guide count quantification.
Step 7: Lentiviral Production and Titer Determination
Package the validated plasmid library into lentiviral particles using HEK293T packaging cells and a three-plasmid system (transfer vector + psPAX2 packaging plasmid + pMD2.G envelope plasmid). Use endotoxin-free plasmid preparations for transfection.
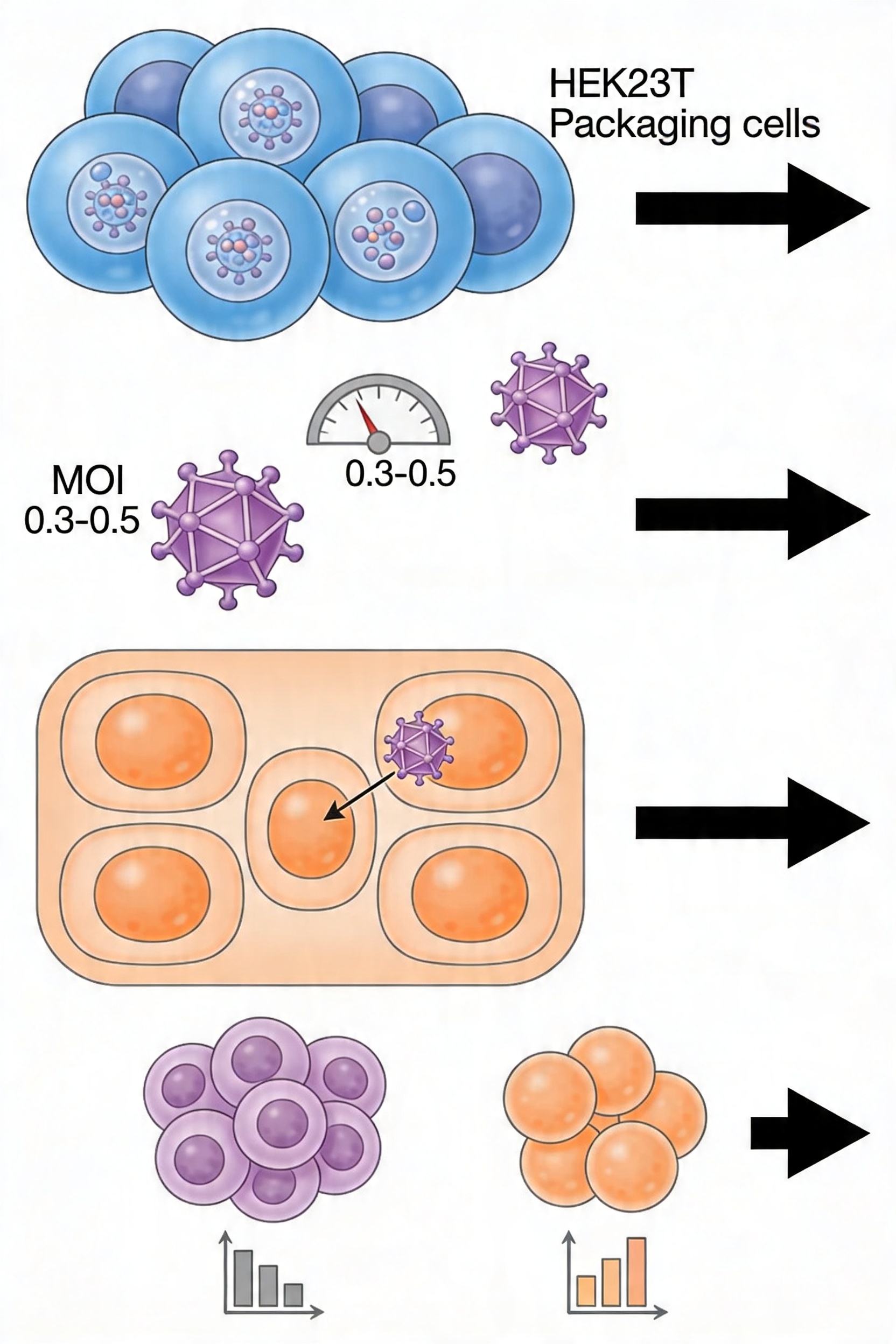
Target viral titer ≥10⁶ transducing units per milliliter (TU/mL). The target multiplicity of infection (MOI) for pooled screens is 0.3 to 0.5, which under Poisson statistics ensures that approximately 74–79% of transduced cells received a single viral copy. At MOI 0.3, approximately 70% of cells will be uninfected, 26% singly infected, and 4% multiply infected.
Step 8: Screen Execution and Coverage Maintenance
Maintaining library coverage throughout the screen — not just at transduction — is the most consistently underappreciated operational requirement in screen design.
A minimum of 300 to 500 cells per guide sequence must be maintained at every passage, selection, or sorting step throughout the screen. For a 100,000-guide library, this means maintaining a minimum of 30–50 million cells at each step. For a 20,000-guide focused library, 6–10 million cells per step. Cell bottlenecks below this threshold introduce stochastic representation losses that manifest as noise in the differential abundance analysis.
This coverage requirement has direct implications for cell culture scale: flask format, split ratios, and the number of cells sorted or selected at each step must all be calculated against the guide count of the library before the screen begins.
Step 9: Genomic DNA Harvest, Library Preparation, and Hit Calling
At screen endpoints, harvest genomic DNA from each population. Amplify the guide insert by PCR using primers flanking the chromosomal integration site (positioned ≥50 bp from the cloning site boundary). Add Illumina sequencing adapters in a secondary PCR and sequence to a depth of at least 200–500 reads per guide per sample.
For differential abundance analysis between conditions, validated computational frameworks include:
- MAGeCK: Negative binomial model; robust to low-count guides; widely used for essential gene screens
- BAGEL: Bayesian framework that incorporates prior knowledge of essential and non-essential gene sets; effective for viability screens
- CRISPRBetaBinomial: Accounts for overdispersion in guide count distributions; appropriate when library representation is variable
For hit validation, clone the top-ranking individual sgRNAs targeting candidate genes into arrayed format and confirm phenotype independently. Indel rate at the cleavage site should be quantified by targeted NGS to establish genotype-phenotype correlation.
Troubleshooting Reference
| Failure Mode |
Root Cause |
Diagnostic |
Mitigation |
| Poor guide representation in plasmid library |
Synthesis bias, excessive PCR cycles, or low transformation coverage |
Compare pre/post-amplification sequencing; check colony count |
Start from high-uniformity pool; reduce to 15 cycles; increase electroporation number |
| High skew ratio (>5) |
Amplification bias or low-complexity starting material |
Sequencing distribution analysis |
Add betaine/DMSO; switch polymerase; increase parallel reaction count |
| Guide dropout at plasmid stage |
Insufficient colony coverage |
Colony count before harvest |
Scale up transformation; use large-format plates; harvest by scraping |
| Multiple integration per cell |
Titer underestimation; MOI too high |
Single-integration PCR assay; antibiotic selection titration |
Re-titer virus; target MOI 0.3–0.5 |
| Coverage bottleneck in screen |
Insufficient cell number at passage/selection |
Track cell count at every step |
Calculate required cell numbers from library size before screen start |
| Off-target artifacts in hit list |
High off-target activity of selected guides |
Check phenotype concordance across multiple guides per gene |
Apply Cas-OFFinder filtering at design stage; require ≥3/6 guides to reproduce |
Dynegene CRISPR Library Platform
Dynegene's CRISPR sgRNA Library service provides synthesis support from guide sequence pool through cloned library construction. The synthesis platform supports:
- Library complexity up to 4.35 million unique sequences per chip
- Oligo lengths from 2 to 350 nt — accommodating standard 80–130 nt library oligos and extended architectures for combinatorial and base editing libraries
- Standard turnaround of 1 week for oligos up to 150 nt (covers the large majority of sgRNA library designs), 2 weeks for 151–230 nt, and 3 weeks for 231–350 nt
- High accuracy and uniformity as core platform performance specifications, which is the synthesis-layer determinant of downstream library quality
For teams building combinatorial dual-guide libraries, base editing libraries, or prime editing pegRNA libraries, Dynegene's Variant Library service provides complementary synthesis capability for these extended guide architectures.
The Oligo Pools service serves as the synthesis foundation for programs that require the full oligo pool format rather than the cloned library delivery, including teams that perform in-house cloning into custom vectors. For downstream library construction reagents including high-efficiency cloning tools, Dynegene's QuarCam Seamless Cloning Kit supports Gibson Assembly-based library construction workflows.
For detailed synthesis specifications, quality documentation, and ordering guidance, the Technical Resources and FAQ pages address platform-specific questions for CRISPR library projects. Published validation data for Dynegene's CRISPR library synthesis platform is available through the Citation and References section.
 NGSHybridization Capture Probe NGS custom probes MRD custom probes QuarStar Human MethylCap Panel QuarStar Liquid Pan-Cancer Panel 3.0 QuarStar Pan-Cancer Lite Panel 3.0 QuarStar Pan-Cancer Fusion Panel 1.0 QuarStar Pan Cancer Panel 1.0 QuarStar Pan-Cancer Panel 1000-Gene QuarStar HLA Panel QuarXeq HRD panel QuarXeq Mitochondrial Probes Whole Exome Sequencing Probes QuarStar Human All Exon Probes 4.0 (Heredity) QuarStar Human All Exon Probes 4.0 (Tumor) QuarStar Human All Exon Probes 4.0 (Standard) QuarXeq Human All Exon Probes 1.0 (RNA) QuarXeq Human All Exon Probes 3.0 (RNA) Library Preparation DNA Library Preparation Kit Fragmentation Reagent mRNA Capture Kit rRNA Depletion Kit QuarPro Superfast T4 DNA Ligase Dynegene Adapter Family Hybridization Capture QuarHyb Super DNA Reagent Kit QuarHyb DNA Plus3 Reagent Kit QuarHyb DNA Reagent Kit Plus QuarHyb One Reagent Kit QuarHyb Super Reagent Kit Pro Dynegene Blocker Family QuarHyb DNA Reagent Kit Pro(Methyl) Multiplex PCR QuarMultiple BRCA Amplicon QuarMultiple PCR Capture Kit 2.0 PathoSeq 450 Pathogen Library Corollary Reagent Streptavidin magnetic beads Equipment and Software The iQuars50 NGS Prep System
NGSHybridization Capture Probe NGS custom probes MRD custom probes QuarStar Human MethylCap Panel QuarStar Liquid Pan-Cancer Panel 3.0 QuarStar Pan-Cancer Lite Panel 3.0 QuarStar Pan-Cancer Fusion Panel 1.0 QuarStar Pan Cancer Panel 1.0 QuarStar Pan-Cancer Panel 1000-Gene QuarStar HLA Panel QuarXeq HRD panel QuarXeq Mitochondrial Probes Whole Exome Sequencing Probes QuarStar Human All Exon Probes 4.0 (Heredity) QuarStar Human All Exon Probes 4.0 (Tumor) QuarStar Human All Exon Probes 4.0 (Standard) QuarXeq Human All Exon Probes 1.0 (RNA) QuarXeq Human All Exon Probes 3.0 (RNA) Library Preparation DNA Library Preparation Kit Fragmentation Reagent mRNA Capture Kit rRNA Depletion Kit QuarPro Superfast T4 DNA Ligase Dynegene Adapter Family Hybridization Capture QuarHyb Super DNA Reagent Kit QuarHyb DNA Plus3 Reagent Kit QuarHyb DNA Reagent Kit Plus QuarHyb One Reagent Kit QuarHyb Super Reagent Kit Pro Dynegene Blocker Family QuarHyb DNA Reagent Kit Pro(Methyl) Multiplex PCR QuarMultiple BRCA Amplicon QuarMultiple PCR Capture Kit 2.0 PathoSeq 450 Pathogen Library Corollary Reagent Streptavidin magnetic beads Equipment and Software The iQuars50 NGS Prep System Primers and Probes
Primers and Probes RNA SynthesissgRNA miRNA siRNA
RNA SynthesissgRNA miRNA siRNA Gene
Gene Oligo Pools
Oligo Pools CRISPR sgRNA Library
CRISPR sgRNA Library Antibody Library
Antibody Library Variant Library
Variant Library




 Tel: 400-017-9077
Tel: 400-017-9077 Address: Floor 2, Building 5, No. 248 Guanghua Road, Minhang District, Shanghai
Address: Floor 2, Building 5, No. 248 Guanghua Road, Minhang District, Shanghai Email:
Email: Tel: 400-017-9077
Tel: 400-017-9077 Address: Floor 2, Building 5, No. 248 Guanghua Road, Minhang District, Shanghai
Address: Floor 2, Building 5, No. 248 Guanghua Road, Minhang District, Shanghai Email:
Email: 







